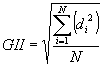Check the bond valence sums of your
inorganic crystal structure
SHELX file input page
Extracted information page
Bond valence results page
CIF-to-SHELX-conversion
SHELX file input page
The "Test your SHELX file" page expects inorganic crystal structure data in the SHELX format as its input. It is assumed that you are familiar with the syntax of SHELX *.ins or *.res files, if not please refer to the SHELX manual (accessible under http://shelx.uni-ac.gwdg.de/SHELX/shelx.pdf ).
The separate "Test your CIF file" page enables you to convert most valid CIF files into a valid SHELX format file suitable for the BV calculations.(For details on the CIF-->SHELX conversion see below).
Copying the information between the horizontal rulers and pasting it into the input field of the "Test your SHELX file" page should produce a valid example file:
TITL 79482-ICSD in space group P 42/m n m
CELL 0.71073 8.2538 8.2538 10.7553 90.000 90.000 90.000
ZERR 4 0.0004 0.0004 0.0006 0.001 0.001 0.001
LATT 1
SYMM - X, - Y, + Z
SYMM 1/2 + X, 1/2 - Y, 1/2 + Z
SYMM 1/2 - X, 1/2 + Y, 1/2 + Z
SYMM + Y, + X, + Z
SYMM - Y, - X, + Z
SYMM 1/2 - Y, 1/2 + X, 1/2 + Z
SYMM 1/2 + Y, 1/2 - X, 1/2 + Z
SFAC K+1 Ni+2 P+5 S-2
UNIT 4 4 4 16
FVAR 1.0
Ni1 2 0.00000 0.00000 0.00000 10.12500 0.02020 0.02020 =
0.02600 0.00000 0.00000 -0.00840
Ni2 2 0.00000 0.00000 0.50000 10.12500 0.02180 0.02180 =
0.02600 0.00000 0.00000 0.00980
S1 4 0.36350 0.36350 0.14420 10.50000 0.03450 0.03450 =
0.02400 -0.00070 -0.00070 -0.01710
S2 4 0.00430 0.26790 0.00000 10.50000 0.02440 0.02150 =
0.04170 0.00000 0.00000 -0.00690
P1 3 0.25060 0.25060 0.00000 10.25000 0.02470 0.02470 =
0.02770 0.00000 0.00000 -0.01140
K1 1 0.00000 0.50000 0.25000 10.25000 0.04590 0.04590 =
0.03200 0.00000 0.00000 0.00000
HKLF 4
END
Only a part of the entries (shown in red) is effectively required and processed. The program just ignores most other SHELX command lines as well as comment lines.
You will have to ensure that all the atom types are specified in a single SFAC line, where also the probable oxidation state of each atom needs to be specified (e.g. as Ni+2 or S-2). Atom types specified by the element name only will be interpreted as neutral atoms.
The determination of Hydrogen positions from X-ray data necessarily leads to an underestimation of the H-X bond length. Replacing the "H" atoms in structure data files by the pseudoatom "HX" (both in SFAC line and in the the atom names of the atomic position lines) should approximately compensate this effect and thus makes it possible to judge the plausiblility of X-ray structure determinations, where all Hydrogen positions have been refined. It should still be kept in mind that the refined H position from XRD structure determinations does not coincide with the position of the nucleus of the H atom.
As a large number of structures contains Ammonium ions NH4+ for which in most cases only the position of the center of gravity is known, a further pseudoatom "NH" has been included in the cation list. Ammonium cations have to be addressed as "NH+1" in the SFAC list and the corresponding atom names have to start by "NH" (e.g. NH7 ).
Known inconveniences include:
Origin shifts by a MOVE command are ignored. (A warning is printed.)
The use of free variables listed in a FVAR command is not yet implemented. These parameters have to be translated into explicit parameters (positions, occupancies) for the respective atoms.
The atom name has to start with the full element symbol (e.g. "Lead1" or "P7" are not valid atom names for a Pb atom).
Fractional oxidation states are not permitted as part of the SHELX input file. You may of course emulate them by two or more atoms of different types with partial occupancies. Another simpler (and less accurate) workaround is to choose the closest integer value for the SFAC entry and to replace the corresponding value in the "Oxidation states for each atom" line on the "extracted information" page. The program will then use the bond valence parameter set for the chosen integer value, but compare the bond valence sum to the fractional expected valence sum in the calculation of BVS deviations and GII (see below).
The maximum number of crystallographically independent atoms is 70 (and among them < 50 cations and <50 anions), to limit the consumption of server resources by a single job. Tests of larger structures are available by sending the SHELX format file via E-mail to the author.
The bond valence approach fundamentally assumes that bonding occurs only between atoms of different electronegativity. Thus the program is not too useful to check the plausibility of structures containing bonds between like atoms (eg. C-C, S-S, Hg-Hg, Mo-Mo ...). As an exception from that rule e.g. thiosulfates can be BV-checked assigning oxidation states -2 to the terminal S and +6 to the central S of the S2O32--group.
After pressing the "PREPARE INPUT FILE FOR BV CALCULATIONS" button below the input area, the program tries to extract the required data from the submitted SHELX file. These data are listed on the
Extracted information page
Before starting the actual bond valence tests by pressing the "START BV TEST" button at the bottom of this page you should browse through the page to check, whether the extracted information looks plausible.
If the bond valence parameters for a cation-anion pair are not available in the database a corresponding error message is printed in bold. The parameters for this pair are then set to R0=0, b=1, R(cutoff)=1 which results in zero bond valences irrespective of the bond distance.
If you know about useful parameters for this atom pair, you may edit the corresponding entries in the BV parameter list below the error message. Otherwise the bond valences calculated in the next step will be only of limited usefulness.
For a few atom pairs BV(M-X) parameters are extrapolated from interactions of the same cation M to other types of anions. (The extrapolated parameters are not published in the softBV database.) A warning is printed if one these parameters is used, since the accuracy of all these parameters is probably lower than for the BV parameters that have been directly determined from experimental data. Special care has to be taken if such an extrapolated parameter set is required to model the strong interactions in the first coordination shell.
For compounds that contain different types of "anions" (i.e. different types of elements with negative oxidation states) the effectively used bond valence parameter b will differ from the tabulated softBV values to account for the mutual equalization of bond softnesses. For each cation the program calculates a weighted mean BVP baverage(cation) averaging over all anion types [weigthing factor = number of these anions per unit cell * abs(oxidation state)] and calculates the effective b-value
beffective(cation,anion) = f* baverage(cation) + (1 - f ) * b(cation,anion),
with an empirical factor f = 2/3. The values of beffective are listed at the end of the results page if they differ from the tabulated values b(cation,anion). This averaging becomes relevant for compounds that contain anions with strongly different softnesses. It should be noted that compounds with containing different types of anions had been excluded from the reference data sets for the determination of softBV parameters (while most compounds in the reference data sets contained different types of cations).
If the "required bond valence parameters" frame is completely empty, the most probable error is that you did not specify the oxidation states of the atoms in the SFAC line of the SHELX file.
An error message "n(AtomX): #.# occupancy >>1 makes no sense!" is typically related to the special meaning of "occupancies" in the SHELX input file (occupancy multiplied by the ratio between the multiplicity of this site and the multiplicity of the general site for this space group). This is not taken into account by some file format conversion programs.
The program also checks the charge balance and prints a warning "!!Sum of charges per formula unit #.###!!!! Unbalanced charges: check occupancies and oxidation states (<0 for anions)" if a significant deviation from neutrality is noted.
Bond valence results page
For each atom the bond valence sums (BVS) as well as the bond lengths and bond valences of all bonds that contribute at least s=0.04 valence units to it are listed. Please note that the bond valence sum often exceeds the sum of the individually displayed bond valences as the BVS includes all contributions up to the maximum bond distance R(cut-off) specified in the database for that atom pair.
Deviations of the bond valence sums are displayed for each atom. The "global instability index" GII as defined by Salinas-Sanchez et al (1992):
is calculated as a measure for the plausibility of the structure, where di represents the difference between the formal oxidation state for atom i and its bond valence sum, while N is the number of atoms in the asymmetric unit. The GII value is often below 0.1 valence units (v.u.) for unstrained structures and as large as 0.2 v.u. in a structure with lattice induced strains. Crystal structures with a GII greater than 0.2 v.u. are typically found to be unstable, and reports of such structures are in most cases found to be incorrect. Examples of GII values (based on softBV and conventional parameters) for recently published structures are found
here.
A common source of high GII values are missing atoms or a low quality determinations of H positions which are almost inevitable from X-ray data. (In this case you may replace the "H" atoms by the psudoatom "HX" to check whether your XRD structure determination is in line with the commonly found apparent shortening of the H-X bond.)
Please keep also in mind that the GII is strongly affected by the choice of the formal oxidation state. So maybe you just have to correct the assignment of oxidation states.
If some of the atom positions are only partially occupied this is taken into account by scaling the bond valence by the fractional occupancy of the bonding partner, but not by the fractional occupancy of the central ion. This handling of partial occupancies implicitly assumes that the occupancy of one site does not affect the occupancy probability of a counterain at a neighboring site, which is surely not always the case. (Here in contrast to the SHELX format input file an occupancy of 1.0 means a fully occupied site independent of its site symmetry.)
Since the GII only judges the plausibility of cation-anion distances, unusually short cation-cation or anion-anion distances are listed together with their occupancy sums as an additional plausibility test. The expected minimum distances are based on crude empirical exclusion radii. Violated expected minimum distances are shown only if the sum of the occupancies exceeds 1.0.
CIF-to-SHELX-conversion
The "Test your CIF file" page enables you to convert most valid CIF files that contain the necessary information into valid SHELX format files suitable for the BV calculations. This CIF --> SHELX file converter is still a test version, please report unexpected results to the author.
The symmetry is derived (preferably) from the Hall space group symbol [Hall, S. R. (1981). Acta Cryst. A37, 517-525] or if this should be missing from the Hermann-Mauguin space group symbol (but so far not from the explicit x,y,z symmetry cards). Thus it is essential that the information in the "_symmetry_space_group_name_Hall" or "_symmetry_space_group_name_H-M" is correctly provided in the CIF file. The Hall symbol is checked first as it gives the space-group setting explicitly.
A remark line is added to list the identified space group and the source of the interpreted information. A few uncommon settings have not yet been implemented.
According to the CIF dictionary a bar above a symmetry axis should be translated into a "-" sign preceding the
the number(e.g. 'P -4 3 n', not 'P 4- 3 n")
Symbols referring to different axes should be separated from each other by spaces. (The program tries to locate missing spaces but this might fail.)
Examples of valid Hall space groups are: '-P 2ac 2n', '-R 3 2"', 'P 61 2 2 (0 0 -1)'
Examples of valid H-M space groups are: 'P 1 21/m 1', 'P 2/n 2/n 2/n:1', 'R -3 2/m'
For HM space groups it is advisable
- to use the full symbol from International Tables for Crystallography, Vol. A (1987) for monoclinic space groups, but the short symbol for higher symmetries. (The program tries to identify the setting from short symbols for monoclinic space groups, but this might fail.)
- to specify that the rhombohedral setting of rhombohedral space groups should be used by appending ":R" or " R" to the H-M space group symbol ('R -3 2/m:R' or 'R -3 2/m R' correponds to a rhombohedral setting, while 'R -3 2/m' or 'R -3 2/m:H' is interpreted as a hexagonal setting)
-
to specify the choice of the origin in orthogonal space groups if there are 2 choices listed in the International Tables, Vol. A by appending
":1" for the first setting (origin not on center of symmetry)
":2" for the second setting (origin on center of symmetry. This is the default.)
In CIF files extracted from the ICSD database " Z" will be interpreted as the default setting (":2"), while " S" is identified with ":1".
As the CIF file format is very flexible, the author of a CIF file often has various options whether and how to enter a specific information and even published CIF files often deviate from the rules given in the CIF dictionary. The present converter tries to extract as much of the relevant information as possible. Nevertheless it is always necessary to check the plausibility of the results.
CIF files may contain more than one structure data set. All the "data_" flags are listed and the interpreted data section (= the first section or the second if the first does not contain atomic coordinates) is named in the header of the "Converted SHELX file" page. Any text preceding the first "data_" instruction or after the end of the interpreted section is ignored. If you should wish to extract information from other data sections you should go back to the CIF input form and comment out preceding sections by placing a "#" (or any character except spaces or tabs) in front of "data_something" section headers to be ignored. As each data section should contain all the required information, you may as well cut the unnecessary sections from the file.
The converter tries to identify the atom types and formal oxidation states from "_atom_type_symbol" or "_atom_site_type_symbol". This often fails due to lack of information in the CIF files. The program will nevertheless continue (sometimes assuming default values for the oxidation states) as this information can often easily be added or corrected manually. Before proceding to the BV calculations you need to check that the oxidation states are correct and that the atom type numbers given in the atoms lines match the SFAC entries.
Temperatures listed in the SHELX file are extracted from "_diffrn_ambient_temperature" and/or "_cell_measurement_temperature". Especially a value of 20 deg Celsius (the default value in the SHELXL template) should not be taken too serious as it is quite often not adapted by the authors to the effective conditions of the measurement. (Sometimes the correct temperature is stated in the text of the publication)
If the number of formula units Z is not specified in the CIF file, a value of "1" is assumed in the ZERR line and in the calculation of the total number of atoms per unit cell (given in the UNIT line). This information is not required for the BV calculations.
Numerical information on the wavelength used in the diffraction experiment is extracted from the "_diffrn_radiation_wavelength" entry in the CIF file if present. Otherwise the Mo Kalpha wavelength is used as a default value in the CELL line of the output, even if other types of radiation are specified in "_diffrn_radiation_type". This information is not of interest for the BV calculations.
Information on anisotropic atomic displacement factors is so far not extracted as it is not required for the BV calculations.
For informations on recent corrections / updates or any other changes see the history file.